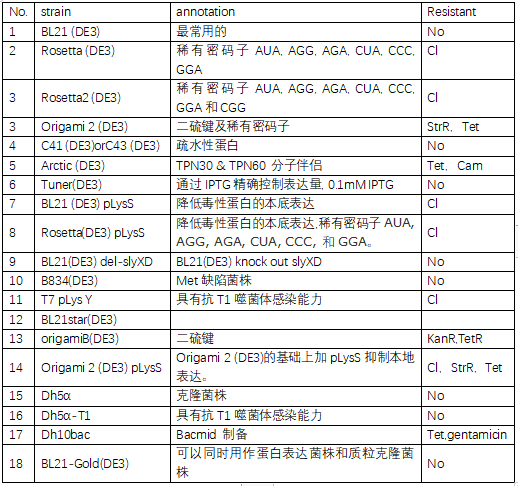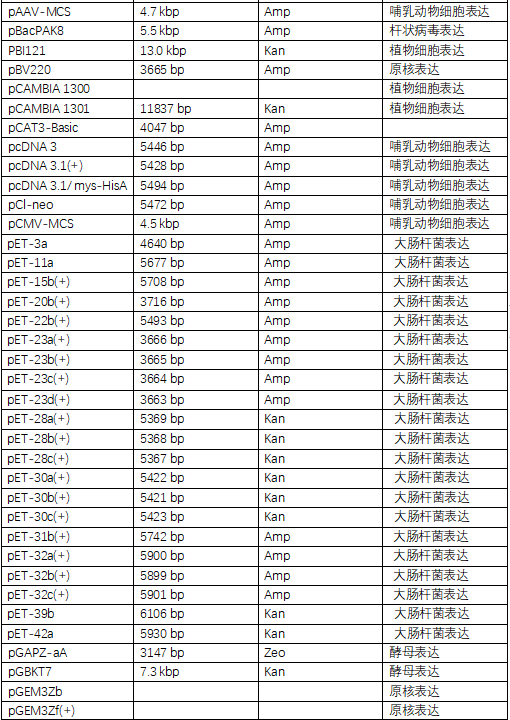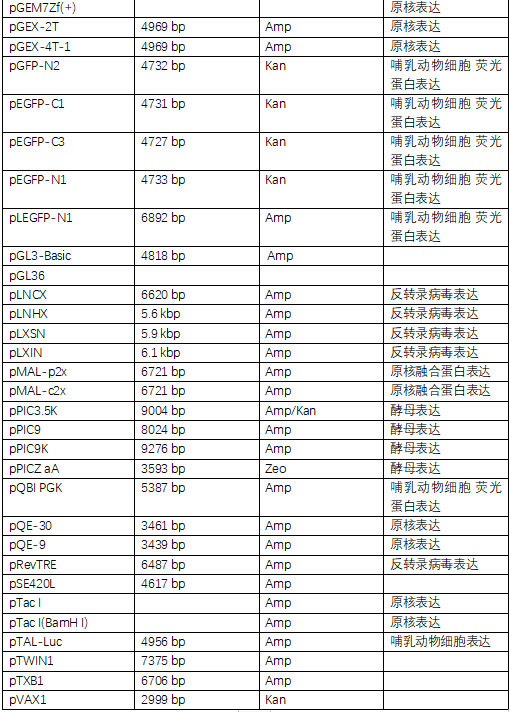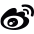



研究一个蛋白质的第一步是拿到它准确的基因序列,有了基因序列,我们就可以构建合适的质粒,表达出所需要的蛋白。
质粒构建,英文名为plasmid construction。选定的目的外源基因(DNA)先要经过PCR扩增。随后用限制性内切酶分别切割载体和外源DNA片段,再使用DNA连接酶将切割后的载体与DNA片段连接,转入宿主细胞。最后经过筛选鉴定出正确的重组克隆质粒,实现目的基因在宿主细胞内的正确表达。
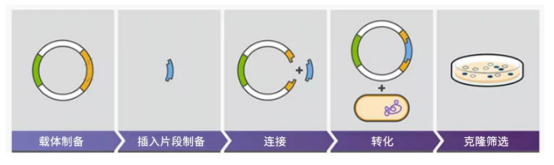
载体通常分为克隆载体(Cloning Vector)与表达载体(Expression Vectors)两种。
克隆载体大多是高拷贝载体,能够将外源基因与克隆载体的质粒连接,导入原核细菌内,进行大量复制克隆。主要用途是保存目的基因片段。
在选择克隆载体时应注意:
①具有自主复制能力,拷贝数高;
②携带易于筛选的选择标记;
③含有多种限制酶的单一识别序列,以供外源基因插入;
④载体应尽可能小(<15kb),便于导入细胞和进行繁殖;
⑤使用安全。克隆载体应只存在于有限范围的宿主内,在宿主体内不进行重组,不发生转移,不产生有害性状,并且不能离开工程宿主自由扩散。
表达载体是为了使插入的外源DNA序列转录、翻译成多肽链而进行特殊设计的克隆载体。它含有特定的表达系统元件,即启动子--核糖体结合位点--克隆位点--转录终止信号。
根据表达的类型,表达载体可分四类:
非融合型表达载体,如PKK223-3;
分泌型表达载体,如PINⅢ-ompA1;
融合蛋白型表达载体,如PGEX;
包涵体型表达载体,如pBV220。
运用PCR扩增目的基因的过程中,引物设计十分关键,以下是需要注意的:
①引物长度最好在18-30bp左右,常用的长度是 20-22bp;
②引物 Tm 值最好在 60℃ 左右,两条引物间 Tm 值要保持接近,相差最好不要超过 5°C;
③GC 的含量标准通常为 40%-60% 或45-55%;
④引物自身不应含有连续超过4个的互补的碱基,避免形成发卡结构或形成引物二聚体;
⑤引物的3' 端要避免出现连续重复的碱基,如 GGG 或 CCC ,这会导致错配的发生,最后一个碱基最好是 G 或 C;
⑥在引物的 5′ 端添加酶切位点时(在不影响扩增的特异性的前提下),根据酶切位点序列,需要添加不同种类和数量的保护碱基,通常多加3个碱基即可满足保护酶切位点的需求;
⑦上下游引物最好加入不同的酶切位点,相同的酶切位点可能导致目的基因片段出现倒置连接现象,影响基因序列的正常表达。
扩增基因的过程并不困难,但未必能保证百分之百的成功率。
如结果出现引物二聚体、非特异性条带(大小不对)的现象,可以通过降低模板和引物的浓度、降低镁离子浓度、适当减少酶量,提高退火温度,来提高扩增特异性。
若出现凝胶条带弥散状态,多为模板不纯、反应体系中各成分比例不合适、退火温度设置偏低、循环次数过多等原因。
长片段基因的扩增容易增加点突变和错配率,选用高扩增能力、高保真、可靠性强的聚合酶也很关键。
在操作酶切连接的步骤时也要定性定量,保证酶活性充足,例如:双酶切尽可能选择同种缓冲buffer,一般用量40U单位以上(用量不超过总体积1/10),保证酶切过程充分;可根据 目标片段量(ng)=(载体量(ng)×目标片段长度(kb))/(载体DNA片段长度(kb))×目标片段和载体的摩尔比(1:3-1:8)计算出目标片段和载体的加入量,提高连接效率。
构建好的载体放入感受态细胞中转化(TOP10、DH5α、BL21等,感受态细胞在使用时尽可能保证新鲜,避免反复冻融,冰上孵育和热激时间应严格控制),经抗性、菌落PCR、酶切、测序等多道程序验证,确定是否转化成功,最终获得正确的重组克隆基因片段。