


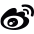



KRAS是癌症中最常见的突变基因之一,G12C突变在非小细胞肺癌中尤为高发。长期以来,KRAS因与核苷酸亲和力极强、缺乏可成药口袋而被视为"不可成药"靶点。
2013年,科学家发现可通过“switch-II 口袋”靶向其非活性状态,开启了 KRAS 靶向药物研发的新方向。虽然目前已经有两款抑制剂(sotorasib 和 adagrasib)获批上市,但临床疗效有限,且耐药性问题逐渐显现,临床上迫切需要新一代更高效的KRAS G12C抑制剂
近日,基因泰克公司的Hans E. Purkey等研究人员报道了一种新型KRAS G12C共价抑制剂Divarasib(GDC-6036)的发现与优化过程。通过增强非共价结合能力,该药物在体外和体内均表现出显著优于现有上市抑制剂的活性和动力学特性,目前已进入临床试验阶段。
相关研究以“Discovery and Characterization of Divarasib (GDC-6036), a Potent Covalent Inhibitor of KRAS G12C”为题,于3月2日在线发表于《Journal of Medicinal Chemistry》上。

研究内容
1. 药物设计与结构优化
KRAS G12C 的 switch-II 口袋包含与Cys12 相邻的近端结合区,以及远端后口袋(back-pocket)。此前的 KRAS G12C 抑制剂研发,多聚焦于优化靶向 Cys12 共价弹头的反应性,而这项研究的核心优化思路是:通过增强与后口袋的相互作用,提升抑制剂与靶点的非共价结合亲和力。
团队选定喹唑啉为母核通过对 C7、C8、C2 位及哌嗪环的分步位点修饰实现活性递进式提升,最终得到候选物 Divarasib。
表1:后口袋优化(R₁取代基的筛选)
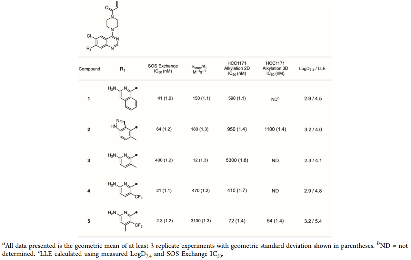
表2. 吡啶5的进一步优化
晶体结构显示,Divarasib较早期化合物向Switch-II螺旋位移约1 Å,back-pocket填充更充分,蛋白-抑制剂接触面显著扩展。
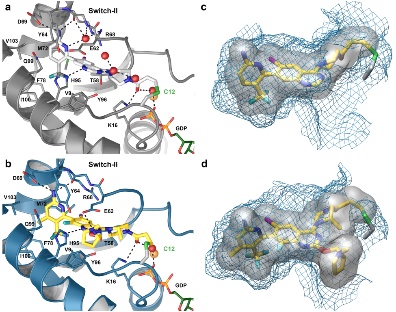
图1. 化合物4和Divarasib(化合物10)与KRAS G12C的共晶结构
2. 机制研究:非共价结合是效力提升的主因
通过动力学实验明确 Divarasib 高活性的核心机制为非共价结合亲和力的大幅提升,而非共价弹头反应性增强。
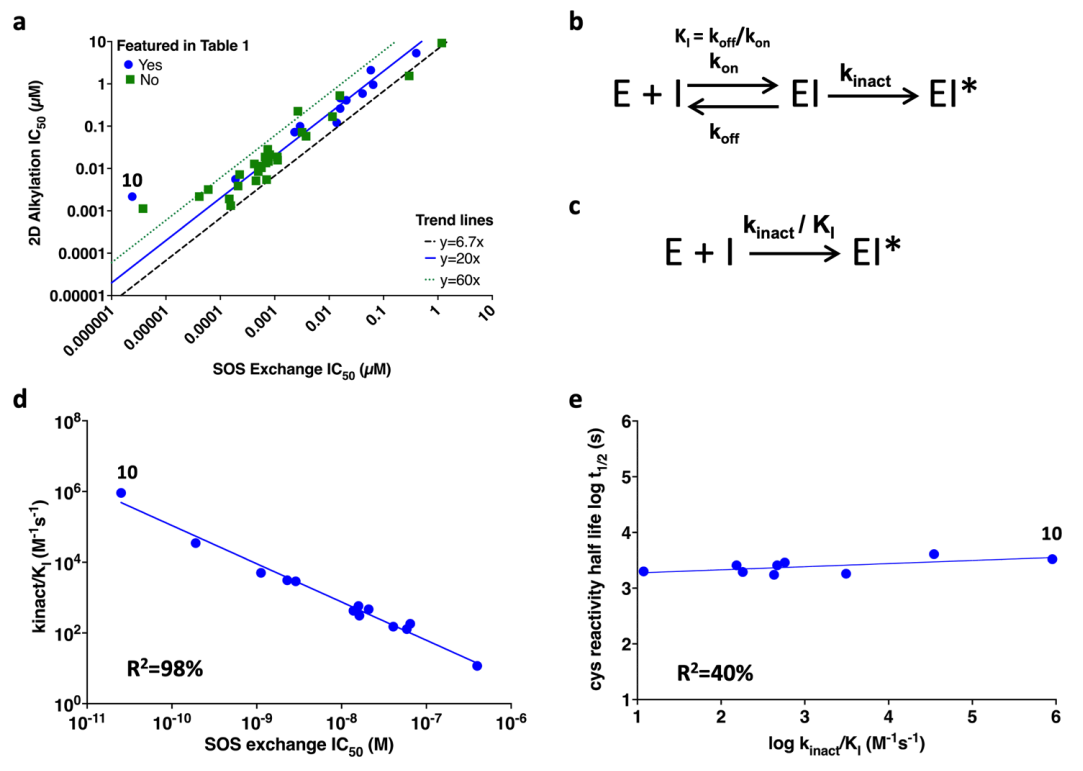
图2. 活性相关性与动力学分析
为验证非共价结合的作用,将丙烯酰胺替换为无反应活性的乙酰胺,SPR实验显示Divarasib衍生物与KRAS G12C 的结合Kd达0.63μM,较早期化合物衍生物提升超800倍,且非共价结合亲和力提升幅度与共价催化效率(Kinact/KI)提升幅度高度匹配。
表3. 选定化合物乙酰胺替代的Kd测定
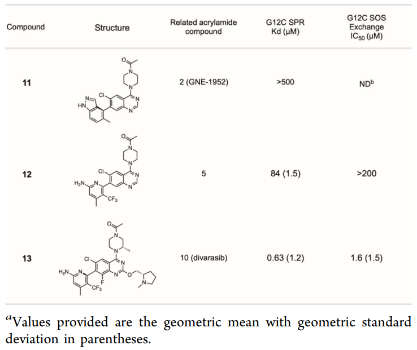
3. Divarasib与已上市G12C抑制剂的比较
Divarasib在生化效力、细胞活性、起效速度、选择性四个维度均显著优于已上市药物。
为更精准评估高活性化合物的靶点结合能力,研究团队从生化效力、细胞活性、起效速度、选择性四个维度,将 Divarasib 与已上市的 sotorasib、adagrasib 进行全面对比,结果显示 Divarasib 在各维度均显著优于两款上市药物。
表4. Divarasib 与已上市KRAS G12C 抑制剂的对比
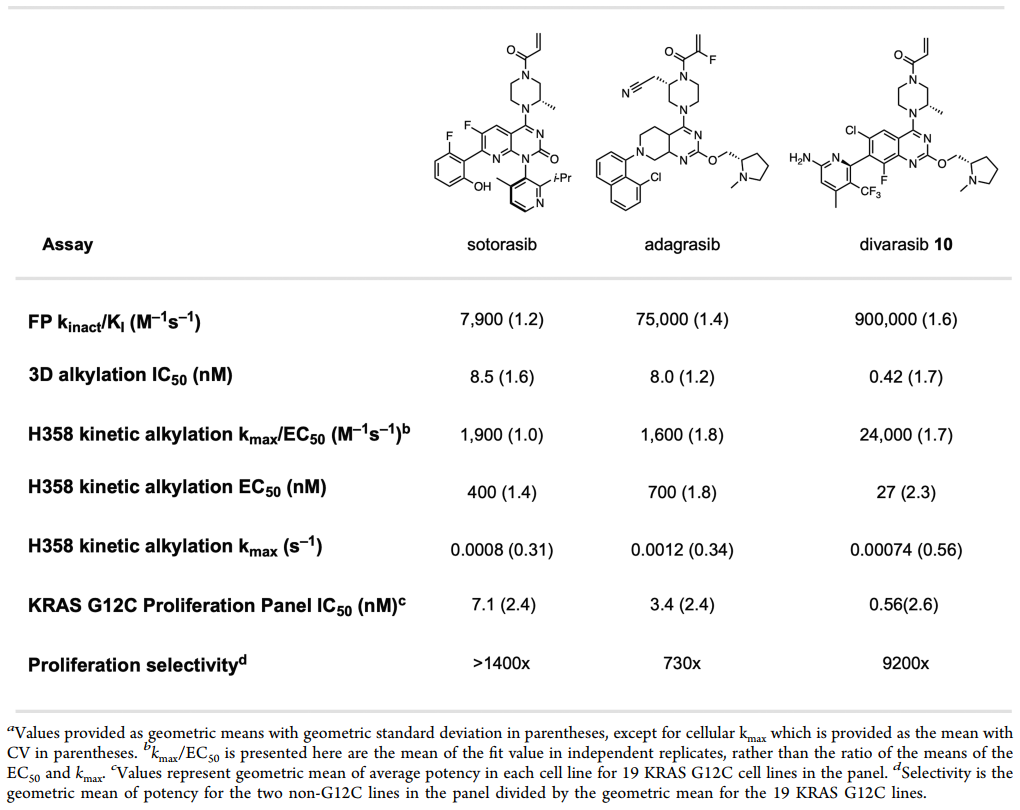
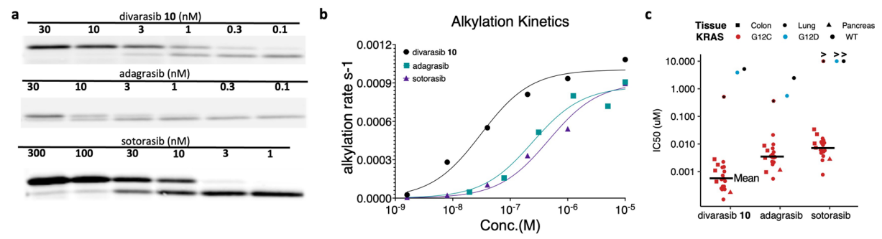
图3. Divarasib与已上市抑制剂的体外细胞活性对比
在超过11,000个半胱氨酸位点中,Divarasib仅对KRAS G12C表现出显著结合,具有极高的靶点选择性
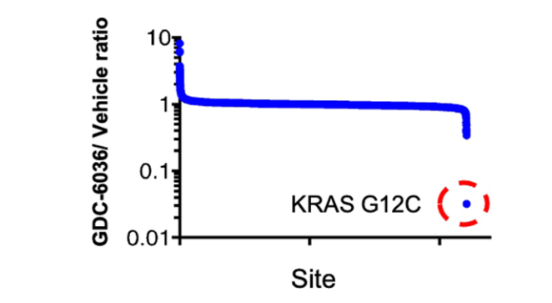
图4. 在蛋白质组学实验中,用10nM 化合物10处理HCC-1171细胞4小时,监测含半胱氨酸肽的损失。
4. 体内药效
在小鼠模型中,Divarasib 在低剂量下即可诱导肿瘤消退,显示出良好的药代动力学特性
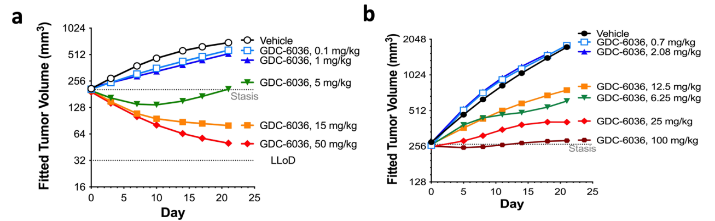
图5. Divarasib 在两种 KRAS G12C 异种移植模型中的体内抗肿瘤活性
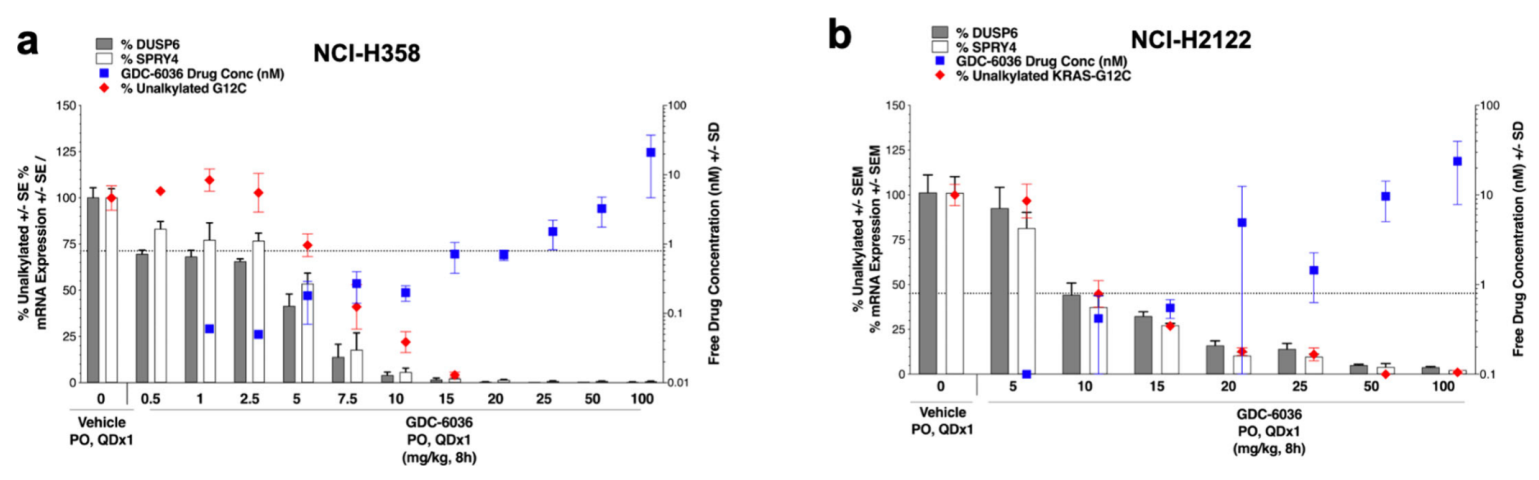
图6. 化合物10的PK/PD关系
总结
本研究是 KRAS G12C 抑制剂研发领域的里程碑研究,首次证实非共价结合亲和力的提升是新一代抑制剂活性突破的核心,并通过精准的结构设计开发出 Divarasib 这一高活性、高选择性的新一代抑制剂。
该研究不仅为 Divarasib 的临床开发奠定了坚实的基础,也为其他 RAS 家族抑制剂(如 KRAS G12D、G12V)的研发提供了重要的设计思路和实验方法,推动了 “不可成药” RAS 靶点的药物研发进程。
同时,Divarasib 的临床潜力已初步显现:临床I期试验中,其单药治疗 KRAS G12C 突变晚期实体瘤,非小细胞肺癌确证缓解率53%、结直肠癌29%;与西妥昔单抗联用时,结直肠癌缓解率飙升至 62%,证实了其联合用药的巨大价值。目前,Divarasib 的多项单药/联合用药临床研究正在开展,覆盖非小细胞肺癌、结直肠癌等多种实体瘤。












